Взаимодействие лекарств имеет место, когда действие одного лекарства изменяется в присутствии другого одновременно применяемого лекарства. Его частота увеличивается с увеличением числа лекарственных средств, включенных в препарат, и продолжительности лечения. Взаимодействие лекарств может происходить до или после всасывания лекарства. Факторы в месте введения лекарства также могут взаимодействовать с препаратом, изменяя его распределение.
Взаимодействие лекарственных препаратов
Взаимодействие лекарственных препаратов происходит до введения или всасывания препарата; может иметь место между двумя лекарствами или лекарством и носителем (растворителем или основой); емкостью (включая систему для внутривенных инфузий) или средой, в которой он вводится (т.е. внутрижелудочную среду). В медицине человека взаимодействие лекарств наиболее часто является результатом добавления лекарственных средств во внутривенные жидкие препараты. В ветеринарии это наиболее часто происходит в условиях интенсивной терапии и реанимации, где часто применяются в/в введение и введение нескольких лекарств. Лекарственная несовместимость может изменять химическую или физическую природу лекарства. Несовместимые реакции могут отражать ухудшение вследствие изменений рН, связывания лекарствами с различными зарядами или иные молекулярные взаимодействия, изменения температуры или воздействие ультрафиолетового излучения.
Внутривенные препараты. Неустойчивые лекарства обычно имеют короткий срок годности в растворе. Восстановленные растворы для парентерального введения всегда должны иметь этикетку с указанием нового срока годности и использоваться в строгом соответствии с указаниями на этикетке продукта после восстановления. Если охлаждение или замораживание указаны на этикетке, они могут увеличить срок годности. Однако рискованно предполагать, что хранение в холодных условиях продлит срок годности лекарства, если его эффективность не была зафиксирована. Замораживание может увеличивать разложение (например, ампициллин), кристаллизацию (например, гепарин, добутамин, фуросемид) или осаждение (например, инсулин) лекарств. Повторное замораживание ранее замороженного и размороженного раствора увеличивает риск потери эффективности. Во избежание инактивации лекарственных средств должна применяться соответствующая восстанавливающая жидкость.
Изменение рН раствора вследствие его неправильного разведения или смешивания с другим лекарством может быть рискованным. Секреция некоторого инсулина зависит от рН, поэтому разведение инсулина раствором, не предусмотренным изготовителем, может изменить pH и, таким образом, скорость секреции инсулина. pH раствора может быть необходимой для поддержания активного лекарства в растворенном или устойчивом состоянии; изменение рН может привести к осаждению или потере устойчивости. Например, кислотонеустойчивые лекарственные вещества (например, пенициллины) могут разрушаться в растворе с низкой рН. Лекарства, приготовленные в виде кислой соли (например, водород хлорид + лидокаин) или в кислых растворах (т.е. гепарин натрий) не должны комбинироваться со щелочными растворами (т.е. натрий бикарбонат). Часто взаимодействие лекарственных препаратов может быть выявлено по визуальному изменению внешнего вида лекарств. Обесцвечивание, мутность и образование осадка являются показателями взаимодействия, в этом случае следует пересмотреть использование лекарства. Если лекарство просто рекристаллизовалось, постепенное подогревание раствора может привести к повторному растворению без потери эффективности. Однако не все взаимодействия будут приводить к физическому изменению внешнего вида. Аналогичным образом изменение физического внешнего вида комбинации лекарств не обязательно указывает на изменение активности лекарства. Например, диазепам смешивался с другими анестетиками, вводимыми при премедикации, без заметного (зафиксированного) изменения эффективности лекарства, несмотря на мутность и обесцвечивание.
В то время как окрашивание дофамина в розовый цвет указывает на инактивирование, изменение цвета добуматина не исключает эффективности, если лекарство используется в течение 24 часов. Приемлемо незначительное окрашивание прокаинамида в желтый цвет; потемнение означает потерю эффективности.
Предосторожность говорит, что любое физическое изменение характера лекарственного средства должно быть причиной не использовать препарат у пациента интенсивной терапии. Лекарства могут связываться и инактивировать друг друга часто вследствие ионного притяжения. Кальций в растворах может вызывать осаждение в сочетании с растворами, содержащими карбонаты (например, бикарбонат натрия). Гепарин несовместим со многими препаратами, такими как аминогликозиды и бета-лактам антибиотики. Таким образом, для сохранения проходимости катетеров, через которые будут вводиться лекарственные вещества, можно использовать раствор натрия хлорида, а не гепарин. В случае присутствия в достаточно высоких концентрациях пенициллины (слабые кислоты) будут связываться с аминогликозидами (слабыми основаниями) и фторхинолонами и инактивировать их.
Некоторые лекарственные препараты могут связываться с емкостями. Например, лекарства, растворимые в липидах (например, диазепам) могут связываться с пластиковыми контейнерами; инсулин связывается с выборочным стеклом и многими пластмассами, в том числе полиэтилен и поливинил; аминогликозиды связываются со стеклом. Связывание с катетерами и капельницами может быть сведено к минимуму промывкой каждой новой системы достаточным объемом раствора (50 мл) до введения лекарства. Лекарства, расфасованные в коричневые бутылки (например, диазепам и фуросемид) защищены от УФ, при перемещении лекарства в другой флакон необходимо сохранить защиту.
Средства местного применения. Взаимодействия лекарственных веществ в средствах местного применения возможно между лекарствами или между лекарствами и раствором, в котором они переносятся. Может оказываться отрицательное воздействие на скорость и степень всасывания вещества. Например, макромолекулярные добавки могут химически связываться с активным лекарственным веществом. Метил, этил, гидроксиэтил и карбоксиметил целлюлоза часто образуют комплексы с лекарствами, которые могут привести к осаждению лекарственного препарата. Растворы часто выбираются вследствие их воздействия на всасывание препарата. Например, замедляющие растворы (полиэтилен гликоль 300) взаимодействуют с лекарством, снижая всасывание, в то время как диметилсульфоксид (DMSO) хорошо известен своей способностью усиливать всасывание многих местно применяемых лекарственных средств.
Фармакокинетическое взаимодействие лекарственных средств
Взаимодействие лекарственных средств, происходящее внутри организма, может быть опасным для жизни. Фармакокинетическое взаимодействие может иметь место, когда одно лекарственное средство изменяет распределение другого. Каждый этап распределения лекарственного средства – всасывание, распределение, метаболизм или выведение – может быть изменен другим лекарственным средством.
Всасывание во рту одного лекарственного средства может быть затруднено вследствие изменений прохождения лекарства через биологические фазы (например, изменения диффузионной способности, скорости растворения и размера частиц перорально вводимых препаратов), изменений местного рН (воздействуя на процент ионизированного и, таким образом, способного рассеиваться лекарства), целостности биологических мембран, регионального кровотока и двигательной активности желудочно-кишечного тракта.
Многие лекарственные вещества препятствуют всасыванию, связывая содержимое полости (взаимодействие лекарственного вещества и рациона); ухудшается всасывание в ротовой полости.
Примерами лекарств, которые будут связывать, и предотвращать всасывание многих лекарств, являются сукральфат, циметидин, гидроксид алюминия и каопектат. Суммарное воздействие продуктов питания на всасывание лекарственного средства зависит от рКа лекарства; от того, является или нет лекарство лабильным к воздействиям рН и ферментов, и места всасывания лекарства (т.е. желудок в сравнении с кишечным трактом). Пища снижает скорость или степень всасывания тетрациклинов (но не доксициклина или миноциклина), фторхинолонов, ампицилина, рифампина, эритромицина (пленочное покрытие) и теофиллина. С другой стороны, всасывание во рту отобранных лекарственных средств (диазепама, гризеофулвина и циклоспорина [в присутствии жира; менее релевантно для микроэмульсии], метапролола, фосфомицина и имидазольных фунгицидов) усиливается в присутствии пищи. Воздействие пищи наиболее важно для антибактериальных средств, зависящих от концентрации (например, фторхинолоны), эффективность которых усиливается высокими концентрациями лекарственного вещества в плазме, лекарств с узким терапевтическим окном и лекарств с крутой кривой зависимости доза-эффект, так как небольшое изменение концентрации лекарства в плазме может вызывать значительные различия к реакции на лекарство. Лекарства, изменяющие двигательную функцию желудка, могут изменять скорость всасывания лекарства во рту.
Так как большинство лекарств всасываются из тонкого кишечника, трудно изменить степень всасывания. Однако возможно изменить скорость всасывания. Введение антихолинергических средств уменьшит опорожнение желудка, увеличивая время до перехода лекарственного средства в тонкий кишечник; могут быть снижены пиковые концентрации лекарства в плазме. В качестве прокинетиков метоклопрамид и цисаприд (или иные прокинетики) должны ускорять опорожнение желудка, что может понижать растворение некоторых лекарственных веществ. Менее вероятно, что увеличение мотильности тонкого кишечника задержит всасывание лекарственных веществ во рту, так как площадь поверхности настолько велика, что этим трудно управлять. Некоторые лекарственные средства изменяют всасывание лекарств, вызывая нарушение всасывания или изменения желудочный кровоток.
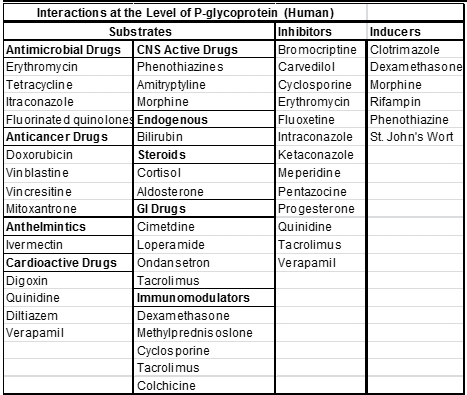
Взаимодействие лекарственных веществ, которое оказывает воздействие на всасывание, также может отражать изменение метаболизма и/или усвоение в энтероците или печени. Лекарства, поступающие в энтероциты, подвержены метаболизму CYP3A, эффлюксу, опосредованному P-gp или прохождению в портальную систему и воздействию метаболической деятельности печени. Цитохром P450 3A является наиболее преобладающим ферментом, метаболизирующим лекарственные средства в клетках кишечника, и опосредует биотрансформацию более половины всех лекарственных средств, имеющихся в настоящее время (для людей). Переносчики, такие как множественная экспортирующая помпа P-gp, облегчают всасывание лекарства или эффлюкс из энтероцита, и лекарственные вещества могут действовать как субстраты или ингибировать, или участвовать в этих белках. Оба белка (CYP 3A и P-gp) имеют общие субстраты лекарственного средства, и взаимодействие между метаболическими ферментами и переносчиками, как оказывается, препятствует распределению многих пероральных лекарственных средств.
Плохая пероральная биодоступность может отражать согласованное действие кишечных ферментов, метаболизирующих лекарственное вещество, и эффлюксных переносчиков; при этом лекарственные вещества и пищевые составляющие могут участвовать или индуцировать, или ингибировать эти белки. Из потенциальных взаимодействий между лекарственным веществом и питательными компонентами наиболее хорошо описана конкуренция между субстратами за транспортные белки. Например, флавеноиды (присутствующие в грейпфрутах) являются ингибиторами нескольких субстратов P-gp, которые увеличивают риск взаимодействия между питательными веществами или питательными веществами и лекарством в процессе всасывания и распределения.
Распределение. Фармакокинетическое взаимодействие лекарственного вещества, которое изменяет распределение лекарства из центрального отдела в периферические ткани, обычно является результатом конкуренции за места связывания белков между двумя или несколькими одновременно вводимыми лекарствами, хотя также важна измененная тканевая перфузия. Большинство взаимодействий лекарств, включающих в себя связывание белка, связано с конкуренцией за места связывания альбумина, в частности за слабые кислоты, липопротеины, глобулины (увеличение с белками острой фазы) и в меньшей степени слабыми основаниями связывания альбуминов (например, бупивакаин, лидокаин). Лекарственное вещество с самой высокой аффинностью вытесняет лекарство с меньшей аффинностью. Чем выше связывание (например, >80%), тем больше изменение соотношения несвязанного лекарственного вещества с вытеснением.
Нестероидные противовоспалительные вещества (NSAID)обычно более чем на 90% связаны с белком; даже незначительное вытеснение повышает концентрацию. Однако у здоровых животных повышение выведения печенью или почками несвязанного лекарства должно уравновешивать увеличение несвязанного лекарства. Является ли это верным или нет, представляет собой вопрос: в соответствии с вкладышем упаковки одновременное введение цефовецина с другими высоко связанными с белком лекарствами повышает концентрацию последнего. Лекарства, изменяющие кровоток к периферическим органам, могут изменять доставку лекарства. Например, средства снижения пост нагрузки могут увеличивать почечный кровоток и, таким образом, выведение почками дигоксина или других выводимых почками лекарств.
Глюкокортикоиды, введенные до восполнения объема жидкости у гиповолемического пациента, усиливает периферическую вазоконстрикцию и снижает распределение других лекарств.
Редко взаимодействие лекарств происходит в месте тканей: квинидин повышает токсичность дигоксина, так как он вытесняет дигоксин из сердечных тканей; в противоположность этому гипокалемия облегчает связывание дигоксина с сердечной тканью, таким образом усиливая кардиотоксичность дигоксина.
Распределение на тканевом уровне также может отражать конкуренцию между лекарственными веществами или питательными составляющими в отношении P-gp. Этот эффлюксный белок обычно располагается в порталах входа и тканях, недосягаемых для действия цитостатических препаратов.
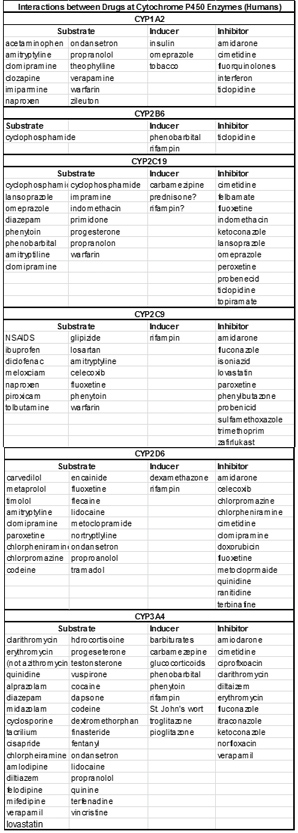
Метаболизм. Фармакокинетическое взаимодействие лекарственного препарата часто изменяет метаболизм одновременно вводимого препарата. При введение препарата, который метаболизируется печенью, целесообразно предвидеть взаимодействие препарата, если добавляется в терапию второе лекарство, которое также метаболизируется печенью. Большинство взаимодействий являются результатом модуляции (фаза I) ферментов печени, метаболизирующих лекарство. Индуцирование ферментов, метаболизирующих лекарство, является защитным механизмом, который облегчает выведение потенциально токсических соединений. Однако индуцирование является палкой о двух концах: хотя стимуляторы обычно повышают уничтожение потенциально токсического лекарства, также может иметь место увеличение образования токсических или канцерогенных метаболитов.
Большинство ферментов CYP являются индуцибильными, хотя реакция на стимуляторы различна в зависимости от вида. Человеческий CYP, на который влияют стимуляторы, включает в себя CYP 1A1/2, 2A6, 2C9, 2C19, 2E1 и 3A4.
Стимуляторы являются субстратами для ферментов; индуцирование обычно зависит от дозы, и часто определяются ферментом, хотя может быть индуцирован боле, чем один CYP. Стимуляторы могут вызывать значительную активность фермента, который в ином случае присутствует в очень низкой концентрации или отсутствует. Максимальная транскрипция в ответ на стимулятор обычно требует от 10 до 12 часов воздействия лекарства, при этом эффект обычно устраняется через 18 часов (в зависимости от дозы) после прекращения воздействия стимулятора. Однако воздействие стимулятора может сохраняться в зависимости от степени деградации CYP. Время, необходимое для того, чтобы CYP достиг новой устойчивой концентрации в ответ на индуцированную скорость синтеза, определяется его скоростью деградации.
Барбитураты считаются стимуляторами CYP. Действительно, наблюдения того, что «толерантность» к гипнотикам, развивающаяся у собак с длительным воздействием барбитуратов, ведет к признанию явлений индуцирования.
Фенобарбитал является одним из наиболее сильных известных микросомальных ферментных стимуляторов и может усиливать гепатотоксичность других гепатотоксических лекарств. Аналогичным образом он увеличивает образование и реакцию на пред лекарства и уменьшает действие себя и других лекарств, метаболизируемых печенью, так как увеличивается выведение этих лекарств.
Семейство CYP2B9, ответственное за метаболизм большого числа лекарств у грызунов, индуцируется фенобарбиталом. Терапевтические дозы фенобарбитала были связаны с индуцированием активности CYPIA. Собаки, страдающие эпилепсией, которые получают лечение фенобарбиталом, могут испытывать первоначальное снижение концентраций через несколько месяцев терапии, несмотря на отсутствие изменения дозы.
Другие противосудорожные лекарственные препараты, которые продемонстрировали подверженность воздействию (снижение концентраций) фенобарбитала, включают в себя зонисамид и леветирацетам.
Также был определен ряд ингибиторов ферментов CYP. Некоторые ингибиторы характеризуются широким ингибированием ферментов, в то время как другие выборочны для отдельного фермента. Очень распространено обратное ингибирование. Наиболее часто (но не всегда), оно проходит после прекращения терапии ингибирования. Подобно индуцированию обратное ингибирование зависит от дозы. Обычно выведение одновременно введенного препарата, метаболизированного печенью, уменьшается с увеличением возможности токсичности или увеличения фармакологической реакции. Кроме того, пред лекарства (например, эналаприл, примидон) имеют меньшую вероятность активирования.
Хлорамфеникол, имидазольные фунгициды и циметидин являются примерами сильных микросомальных ферментных ингибиторов. Одновременное введение с потенциально токсическими препаратами, которые также метаболизируются печенью, следует проводить с осторожностью.
Фторхинолоны, такие как энрофлоксацин и марбофлоксацин, могут повышать концентрации аминофилина в плазме до токсических уровней, предположительно вследствие нарушения выведения теофилина печенью.
Ингибиторный эффект эритромицина может отражать ингибирование CYP 3A.
Хотя азитромицин предположительно обладать меньшей ингибиторной способностью, наша лаборатория зафиксировала заметное увеличение концентраций циклоспорина у пациентов, получающих азитромицин.
Индуцированное лекарственными препаратами ингибирование метаболизма лекарств может использоваться для терапевтической пользы. Циклоспорин и кетаконазол являются субстратами и ингибиторами и P-pg и CYP3A. Соответственно, комбинированное использование этих препаратов может заметно увеличивать период полувыведения и того и другого лекарства.
В нашей лаборатории зафиксирован период полувыведения более 150 часов для циклоспорина (обычно 4-5 часов) у собак, одновременно получающих кетаконазол с целью снижения дозы (и, таким образом, стоимости) CsA при поддержании концентраций циклоспорина.
Циметидин-индуцированное ферментное ингибирование использовалось для предотвращения метаболизма ацетаминофена у кошек с потенциально летальными токсическими метаболитами. Циластатин ингибирует метаболизм имипенема в почечных канальцах; суммарный эффект может продлевать период полувыведения имипенема, но гепато - или ренальная токсичность в результате метаболитов также может сокращаться.
Питание, пол, возраст и другие факторы могут влиять на то, как метаболизирующие лекарственные препараты ферменты могут реагировать на лекарства.
Спирт и 4-метилпиразол конкурентно ингибируют алкогольдегидрогеназу, стабилизирующий лекарственный препарат фермент, который превращает этиленгликоль в его смертельный метаболит.
Выведение. Фармакокинетическое взаимодействие лекарственного вещества может изменять мочевыделение вследствие изменений гломерулярной фильтрации и/или конкуренции между лекарствами за активную канальцевую секрецию. Конкуренция за кислотные, щелочные или органические транспортные белки, ответственная за активную канальцевую секрецию, обычно связана с кислотными лекарствами (бета-лактамы, сульфаниламиды, NSAID, метаболиты метаболизма фазы II, фуросемид), но также может быть связана со щелочными лекарствами (например, прокаинамид, дофамин, триметоприм, опиоды).
Пробенецид все еще иногда используется для продления удаления дорогостоящего пенициллина, так как он конкурирует с пенициллином в отношении транспортного белка.
Лекарства, изменяющие рН мочи и канальцевую ресорбцию, также могут влиять на выведение с мочой. Изменения рН в моче, способствующие образованию большего соотношения ионизированного лекарства (например, кислотный рН мочи и кислотное лекарство) будут способствовать канальцевой реабсорбции лекарства, таким образом, сокращая его выведение и продлевая его период полувыведения. Например, передозировка некоторых лекарств (например, стрихнина) может устраняться ускорением выведения средствами, повышающими кислотность мочи.
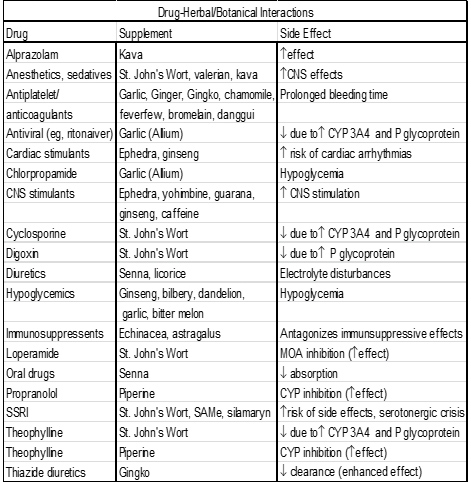
Среди наиболее часто определяемых взаимодействий лекарство - питательное вещество (питательное вещество или добавка), которые изменяют фармакокинетику лекарства, находятся взаимодействия, отражающие метаболизм лекарственного средства, в частности CYP3A4. Известно, что некоторые питательные добавки взаимодействуют с лекарственными средствами. Они включают в себя, в том числе, зверобой (индуцирование CYP3A4, в особенности в кишечнике), эхинацею (индуцирование и ингибирование CYP в кишечнике), гинко билобу (индуцирование CYP219A) и грейпфрут (ингибирование CYP3A4 и ингибирование P-gp).